Initial Areas of Emphasis. Our initial investigations will focus on heart failure and will explore the paradigm that disease pathogenesis is governed by a multi-hit model that can be intervened upon through individualized precision medicine strategies. We postulate that patient specific genomic variants govern disease susceptibility and initiation (primary hit), and that maladaptive inflammatory responses to tissue injury and/or dysfunction (secondary hits) contribute to disease progression. Furthermore, we believe that the development of therapeutics that target each of these mechanisms will revolutionize the care of heart failure patients.
Functional Genomics of Dilated Cardiomyopathy (DCM): Single Cell Mapping of the Human Heart
DCM represents a prominent cause of heart failure in children and adults and is the most common diagnosis in patients listed for heart transplantation. Mutations are increasingly being identified in patients with DCM, a disease previously thought to be idiopathic. Little is known about whether patients who carry mutations in different genes for DCM display distinct disease phenotypes and/or different clinical outcomes including responses to medical therapies.
We believe that DCM should be reclassified based on mutations that give rise to phenotypes. If genetic subtypes of heart failure can be identified based on the mutations found in DCM patients, then unique therapeutic interventions may be designed and used to treat DCM.
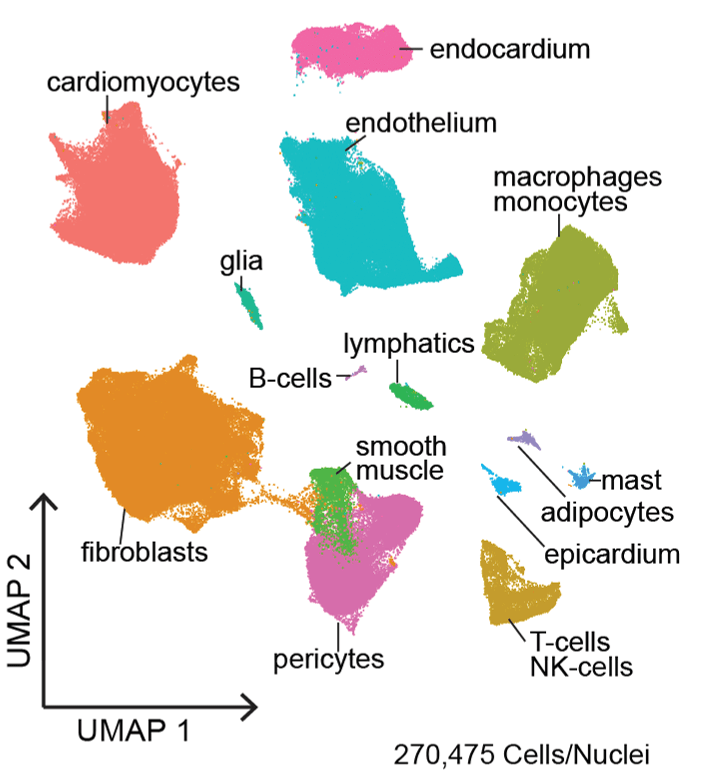
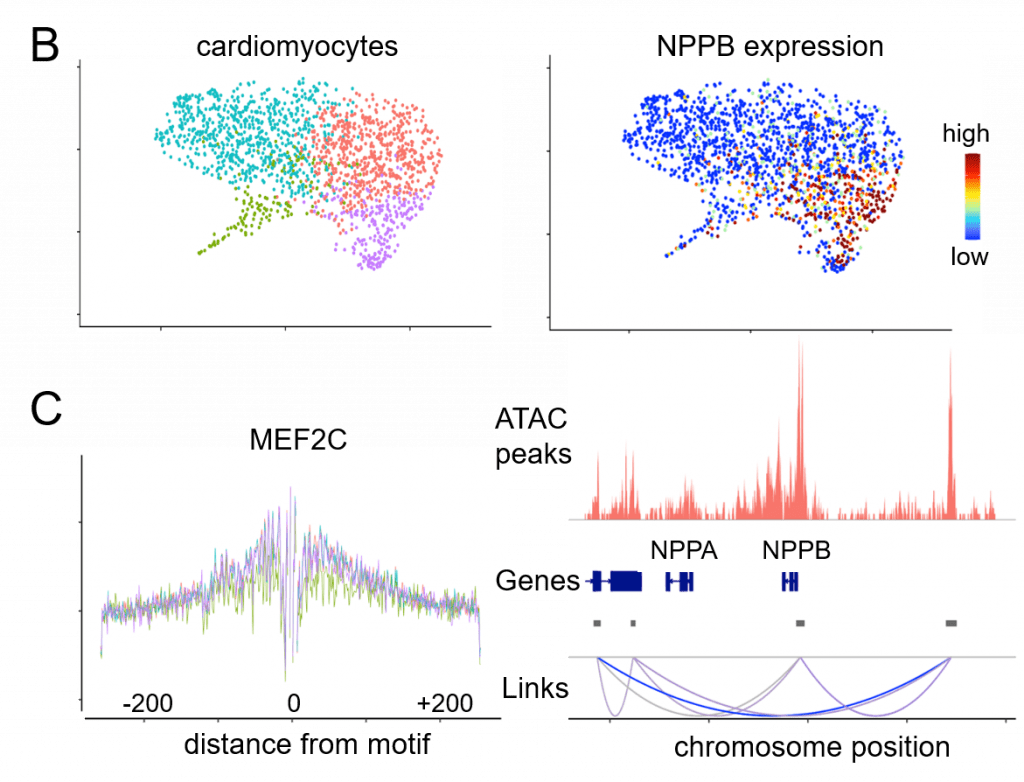
The Washington University Heart Failure Registry (WU-HF Registry) and the Translational Cardiovascular Biobank and Repository (TCBR) are two clinical resources that have been developed to address the genetic subsets of heart failure though phenotypic and pathological analyses. Single nuclei RNA sequencing and single cell RNA sequencing are innovative technologies used within the lab to understand the gene expression of specific cell types in different DCM genotypes.
Novel approaches utilizing engineered heart tissues provide the opportunity to obtain new and unprecedented information into how individual DCM mutations influence mechanical, structural, and electrophysiological properties that contribute to heart failure pathogenesis.
Precision Approaches to Redefine Pediatric and Adult Heart Failure
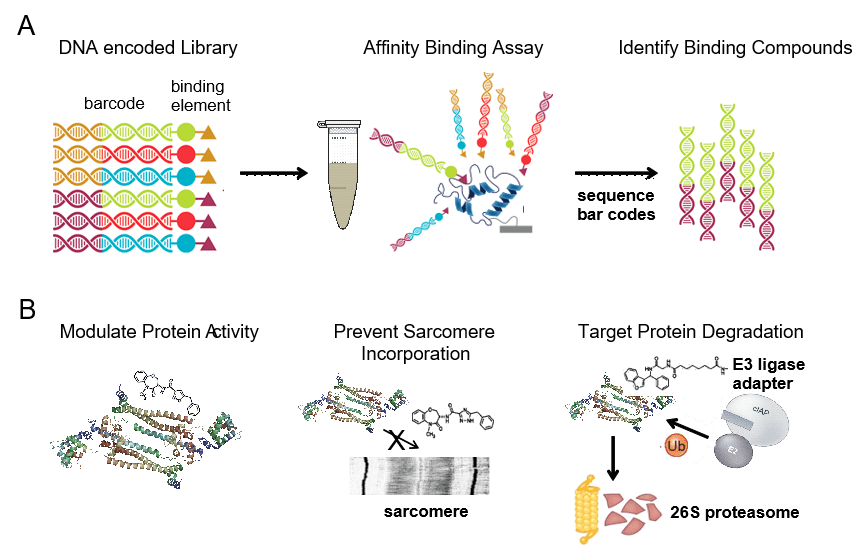
The ultimate goal of CPRi is to develop new therapeutics for DCM. Given that the majority of DCM patients harbor heterozygous missense mutations that display reduced or absent activity, we are identifying compounds that selectively bind to mutant DCM proteins. We use DNA encoded libraries that contain 1000x more entities than traditions small molecule libraries. This technology binds the compounds within the library by affinity allowing to sequence the codes and identify compounds that bind that DCM mutant.
Troponin mutant complexes are used to establish the process, but many DCM variants can be used. The compounds that selectively bind to the DCM mutant proteins ideally will lead to the development of new therapeutics that precisely target the genetic variant of DCM that is being treated.
Targeting inflammation in heart failure (Diagnostics: CCR2 imaging and therapeutics: CCR2 macrophage inhibitors)
Inflammation is considered an important mechanism that contributes to both nonischemic and ischemic cardiomyopathy. In recent years, a renewed interest in targeting the immune system in cardiovascular disease has emerged. This resurgence is powered by the discovery of the cellular mediators of inflammation in the heart and the identification of the mechanisms that orchestrate their activation and damaging effector functions.
We have established that a specific subset of macrophages (identified based on the expression of C-C Chemokine Receptor 2, CCR2) are key mediators of cardiac inflammation and heart failure progression. From a precision medicine perspective, we have defined targetable signaling events that govern the activation of CCR2+ macrophages and successfully designed a molecular imaging probe (compatible with PET/CT or PET/MRI) to visualize CCR2+ macrophages in patients to facilitate the identification of individuals most likely to benefit from therapeutics targeting this cell population.
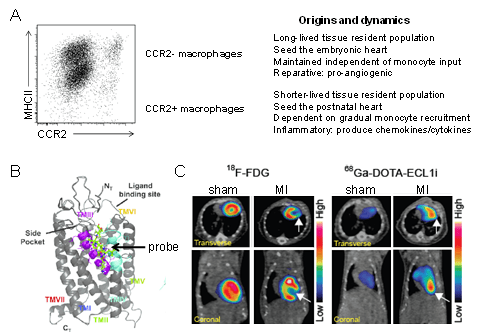
Figure 4. Cardiac macrophage diversity. A ,Flow cytometry plot and schematic depicting the origins, dynamics, and functions of macrophage populations within the heart. B, Model showing that the ECL1i peptide imaging probe binds to CCR2 in an allosteric position. C, PET/CT images demonstrating robust ECL1i tracer uptake within the infarct 4 days following ischemia reperfusion injury. FDG imaging identifies the infarct region.
Therapeutics Targeting CCR2+ Macrophage Activation
Using genetic loss of function models in mice, we have seen that CCR2+ macrophages are activated through concomitant engagement of multiple innate immune receptors including Toll-like receptors (TLRs) and the Interleukin 1 receptor (IL1R) that converge on a signal transduction pathway primarily transduced through MYD88 and NF-kb signaling. We hope to develop therapeutics that target the activation pathway of CCR2+.
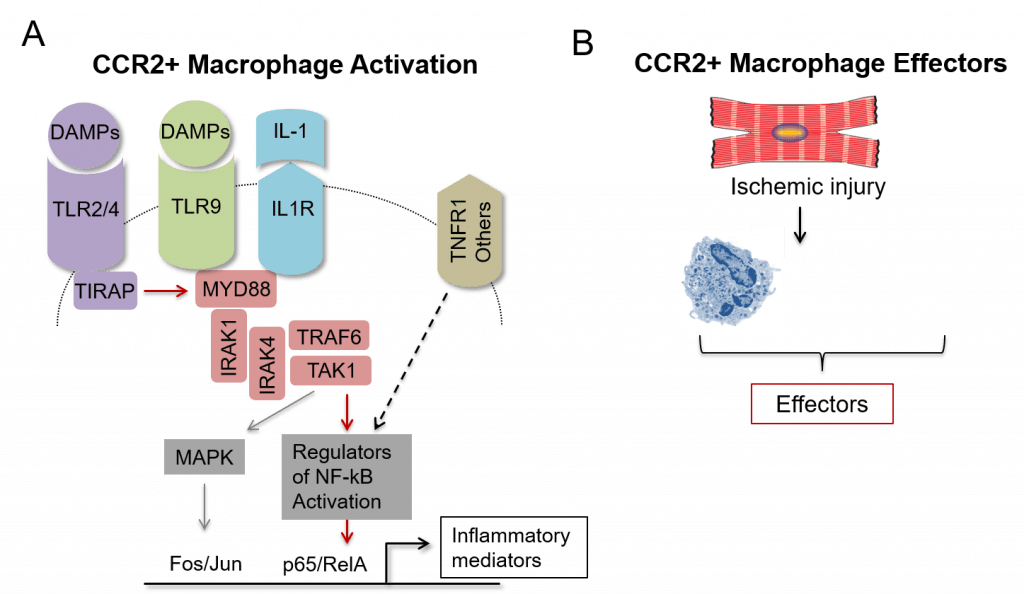
Figure 5. Targeting CCR2+ macrophages. A ,Schematic of the signaling pathway that orchestrates the activation of CCR2+ macrophages. Red arrows denote events required for CCR2+ macrophage activation. B, Diagram highlighting the relevant effectors of CCR2+ macrophages. Red boxes denote the focus of our current studies.
Therapeutics Targeting CCR2+ Macrophage Effector Mechanisms
CCR2+ macrophages exert their effects by generating and secreting chemokines, cytokines, and other effector molecules that promote additional leukocyte recruitment, collateral tissue injury, and adverse remodeling of the myocardium (fibrosis and hypertrophy). To delineate the potential clinical utility of inhibiting the production of these effector molecules, we will study mice that lack expression of IL1b, CCL17, AREG in CCR2+ macrophages using our mouse model of ischemia reperfusion injury.
Therapeutics that Prevent CCR2+ Macrophage Fate Specification
Monocytes display dramatic plasticity and can adopt a vast number of cell fates in response to environmental and cell intrinsic cues. For example, using cell single RNA sequencing, we have discovered that upon entry into the infarcted heart, monocytes differentiate into at least 7 distinct inflammatory and reparative macrophage populations with differing cell morphologies, locations within the infarct, and gene expression profiles (Figure 6A-B).
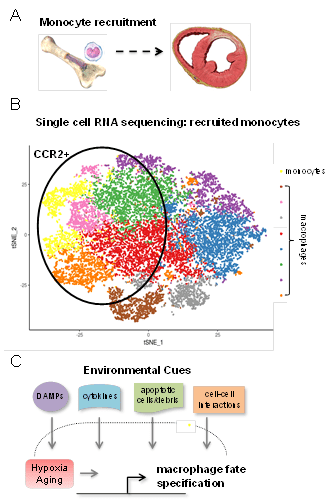
Figure 6. Targeting CCR2+ macrophages. A ,Schematic of the signaling pathway that orchestrates the activation of CCR2+ macrophages. Red arrows denote events required for CCR2+ macrophage activation. B, Diagram highlighting the relevant effectors of CCR2+ macrophages. Red boxes denote the focus of our current studies.
We will utilize mouse models of ischemia reperfusion injury where monocytes are genetically targeted to decipher the impact of environmental and cell intrinsic cues on monocyte fate specification and outcomes following myocardial infarction. A key focus will be to identify the molecular mechanisms that underlie how these instructive cues trigger monocytes to differentiate into CCR2+ macrophages. We envision that such mechanisms would constitute new therapeutic targets to reduce cardiac inflammation (Figure 6C).